
Update on: April 23, 2024
GWAS identify 95 risk loci and provide insights into the neurobiology of post-traumatic stress disorder
Post-traumatic stress disorder (PTSD) genetics are characterized by lower discoverability than most other psychiatric disorders. The contribution to biological understanding from previous genetic studies has thus been limited.
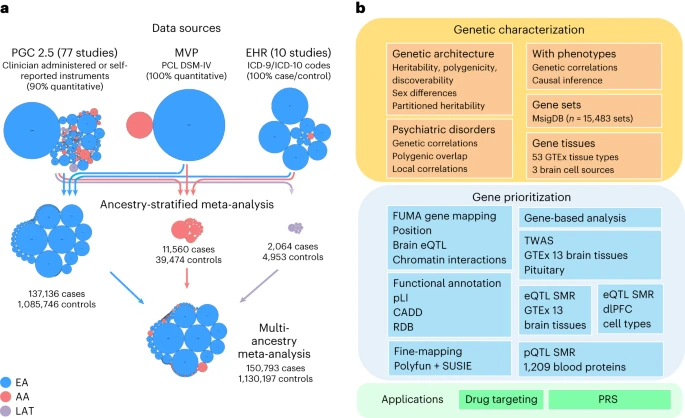
We performed a multi-ancestry meta-analysis of genome-wide association studies across 1,222,882 individuals of European ancestry (137,136 cases) and 58,051 admixed individuals with African and Native American ancestry (13,624 cases). We identified 95 genome-wide significant loci (80 new).
Convergent multi-omic approaches identified 43 potential causal genes, broadly classified as neurotransmitter and ion channel synaptic modulators (for example, GRIA1, GRM8 and CACNA1E), developmental, axon guidance and transcription factors (for example, FOXP2, EFNA5 and DCC), synaptic structure and function genes (for example, PCLO, NCAM1 and PDE4B) and endocrine or immune regulators (for example, ESR1, TRAF3 and TANK). Additional top genes influence stress, immune, fear and threat-related processes, previously hypothesized to underlie PTSD neurobiology. These findings strengthen our understanding of neurobiological systems relevant to PTSD pathophysiology, while also opening new areas for investigation. Reference
A pan-cancer analysis of the microbiome in metastatic cancer
Microbial communities are resident to multiple niches of the human body and are important modulators of the host immune system and responses to anticancer therapies.

Recent studies have shown that complex microbial communities are present within primary tumors. To investigate the presence and relevance of the microbiome in metastases, we integrated mapping and assembly-based metagenomics, genomics, transcriptomics, and clinical data of 4,160 metastatic tumor biopsies. We identified organ-specific tropisms of microbes, enrichments of anaerobic bacteria in hypoxic tumors, associations between microbial diversity and tumor-infiltrating neutrophils, and the association of Fusobacterium with resistance to immune checkpoint blockade (ICB) in lung cancer.
Furthermore, longitudinal tumor sampling revealed temporal evolution of the microbial communities and identified bacteria depleted upon ICB. Together, we generated a pan-cancer resource of the metastatic tumor microbiome that may contribute to advancing treatment strategies. Reference
Single-cell multiomics reveals the interplay of clonal evolution and cellular plasticity in hepatoblastoma
Hepatoblastomas (HB) display heterogeneous cellular phenotypes that influence the clinical outcome, but the underlying mechanisms are poorly understood.
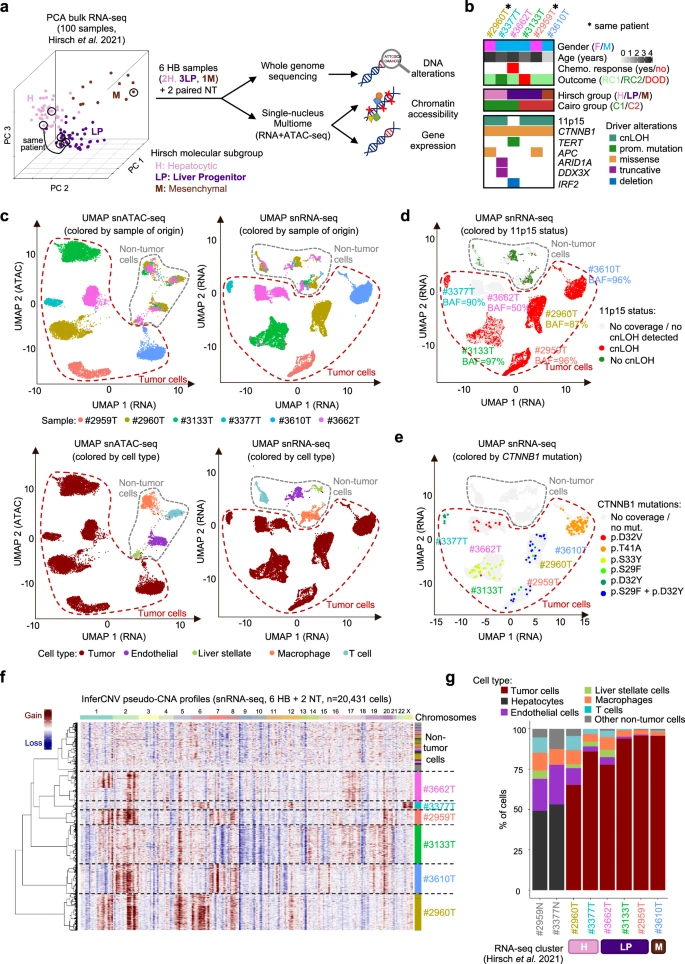
Here, we use a single-cell multiomic strategy to unravel the molecular determinants of this plasticity. We identify a continuum of HB cell states between hepatocytic (scH), liver progenitor (scLP) and mesenchymal (scM) differentiation poles, with an intermediate scH/LP population bordering scLP and scH areas in spatial transcriptomics. Chromatin accessibility landscapes reveal the gene regulatory networks of each differentiation pole, and the sequence of transcription factor activations underlying cell state transitions.
Single-cell mapping of somatic alterations reveals the clonal architecture of each tumor, showing that each genetic subclone displays its own range of cellular plasticity across differentiation states. The most scLP subclones, overexpressing stem cell and DNA repair genes, proliferate faster after neo-adjuvant chemotherapy. These results highlight how the interplay of clonal evolution and epigenetic plasticity shapes the potential of HB subclones to respond to chemotherapy. Reference
Gut microbiome and metabolome profiling in Framingham heart study reveals cholesterol-metabolizing bacteria

Comprehensive WGS analyses provide insights into the genomic architecture of cerebral palsy
We performed whole-genome sequencing (WGS) in 327 children with cerebral palsy (CP) and their biological parents. We classified 37 of 327 (11.3%) children as having pathogenic/likely pathogenic (P/LP) variants and 58 of 327 (17.7%) as having variants of uncertain significance.

Multiple classes of P/LP variants included single-nucleotide variants (SNVs)/indels (6.7%), copy number variations (3.4%) and mitochondrial mutations (1.5%). The COL4A1 gene had the most P/LP SNVs. We also analyzed two pediatric control cohorts (n = 203 trios and n = 89 sib-pair families) to provide a baseline for de novo mutation rates and genetic burden analyses, the latter of which demonstrated associations between de novo deleterious variants and genes related to the nervous system.
An enrichment analysis revealed previously undescribed plausible candidate CP genes (SMOC1, KDM5B, BCL11A and CYP51A1). A multifactorial CP risk profile and substantial presence of P/LP variants combine to support WGS in the diagnostic work-up across all CP and related phenotypes. Reference
Epigenetic variation impacts individual differences in the transcriptional response to influenza infection
Humans display remarkable interindividual variation in their immune response to identical challenges. Yet, our understanding of the genetic and epigenetic factors contributing to such variation remains limited.

Here we performed in-depth genetic, epigenetic and transcriptional profiling on primary macrophages derived from individuals of European and African ancestry before and after infection with influenza A virus.
We show that baseline epigenetic profiles are strongly predictive of the transcriptional response to influenza A virus across individuals. Quantitative trait locus (QTL) mapping revealed highly coordinated genetic effects on gene regulation, with many cis-acting genetic variants impacting concomitantly gene expression and multiple epigenetic marks. These data reveal that ancestry-associated differences in the epigenetic landscape can be genetically controlled, even more than gene expression. Reference
scGIST: gene panel design for spatial transcriptomics with prioritized gene sets
A critical challenge of single-cell spatial transcriptomics (sc-ST) technologies is their panel size.

Being based on fluorescence in situ hybridization, they are typically limited to panels of about a thousand genes. This constrains researchers to build panels from only the marker genes of different cell types and forgo other genes of interest, e.g., genes encoding ligand-receptor complexes or those in specific pathways.
We propose scGIST, a constrained feature selection tool that designs sc-ST panels prioritizing user-specified genes without compromising cell type detection accuracy. We demonstrate scGIST’s efficacy in diverse use cases, highlighting it as a valuable addition to sc-ST’s algorithmic toolbox. Reference
Metabolomic machine learning predictor for diagnosis and prognosis of gastric cancer
Gastric cancer (GC) represents a significant burden of cancer-related mortality worldwide, underscoring an urgent need for the development of early detection strategies and precise postoperative interventions.
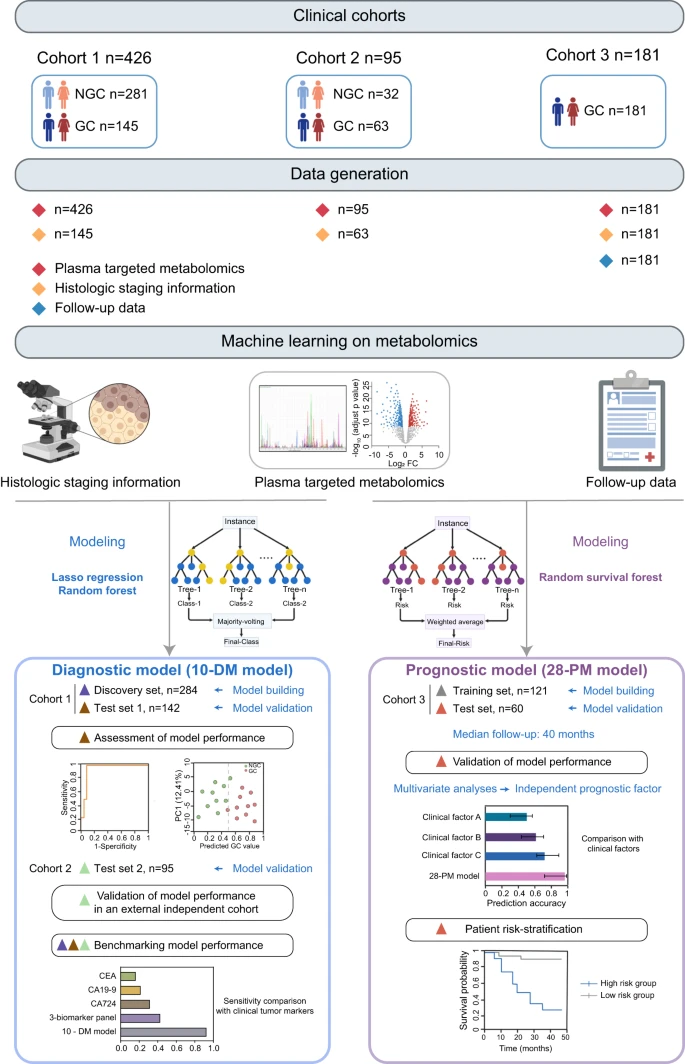
However, the identification of non-invasive biomarkers for early diagnosis and patient risk stratification remains underexplored. Here, we conduct a targeted metabolomics analysis of 702 plasma samples from multi-center participants to elucidate the GC metabolic reprogramming. Our machine learning analysis reveals a 10-metabolite GC diagnostic model, which is validated in an external test set with a sensitivity of 0.905, outperforming conventional methods leveraging cancer protein markers (sensitivity < 0.40).
Additionally, our machine learning-derived prognostic model demonstrates superior performance to traditional models utilizing clinical parameters and effectively stratifies patients into different risk groups to guide precision interventions. Collectively, our findings reveal the metabolic landscape of GC and identify two distinct biomarker panels that enable early detection and prognosis prediction respectively, thus facilitating precision medicine in GC. Reference
Functional dissection of human cardiac enhancers and noncoding de novo variants in congenital heart disease
Rare coding mutations cause ∼45% of congenital heart disease (CHD). Noncoding mutations that perturb cis-regulatory elements (CREs) likely contribute to the remaining cases, but their identification has been problematic.

Using a lentiviral massively parallel reporter assay (lentiMPRA) in human induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs), we functionally evaluated 6,590 noncoding de novo variants (ncDNVs) prioritized from the whole-genome sequencing of 750 CHD trios. A total of 403 ncDNVs substantially affected cardiac CRE activity. A majority increased enhancer activity, often at regions with undetectable reference sequence activity. Of ten DNVs tested by introduction into their native genomic context, four altered the expression of neighboring genes and iPSC-CM transcriptional state.
To prioritize future DNVs for functional testing, we used the MPRA data to develop a regression model, EpiCard. Analysis of an independent CHD cohort by EpiCard found enrichment of DNVs. Together, we developed a scalable system to measure the effect of ncDNVs on CRE activity and deployed it to systematically assess the contribution of ncDNVs to CHD. Reference
Simple but powerful interactive data analysis in R with R/LinekdCharts
In research involving data-rich assays, exploratory data analysis is a crucial step.
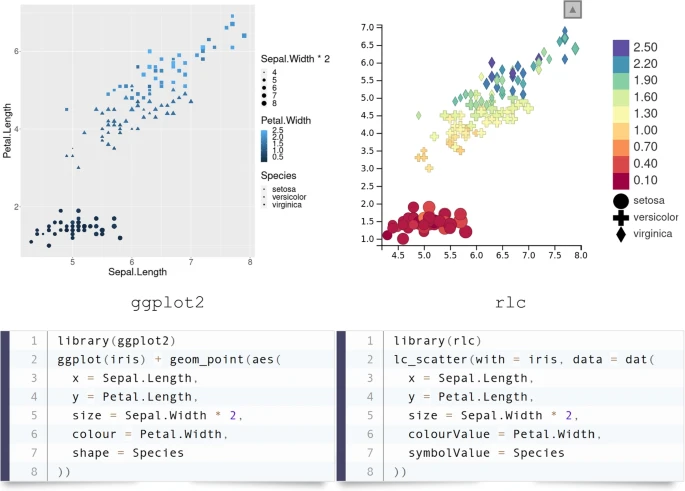
Typically, this involves jumping back and forth between visualizations that provide overview of the whole data and others that dive into details. For example, it might be helpful to have one chart showing a summary statistic for all samples, while a second chart provides details for points selected in the first chart.
We present R/LinkedCharts, a framework that renders this task radically simple, requiring very few lines of code to obtain complex and general visualization, which later can be polished to provide interactive data access of publication quality. Reference
Single-cell multiomics decodes regulatory programs for mouse secondary palate development
Perturbations in gene regulation during palatogenesis can lead to cleft palate, which is among the most common congenital birth defects.
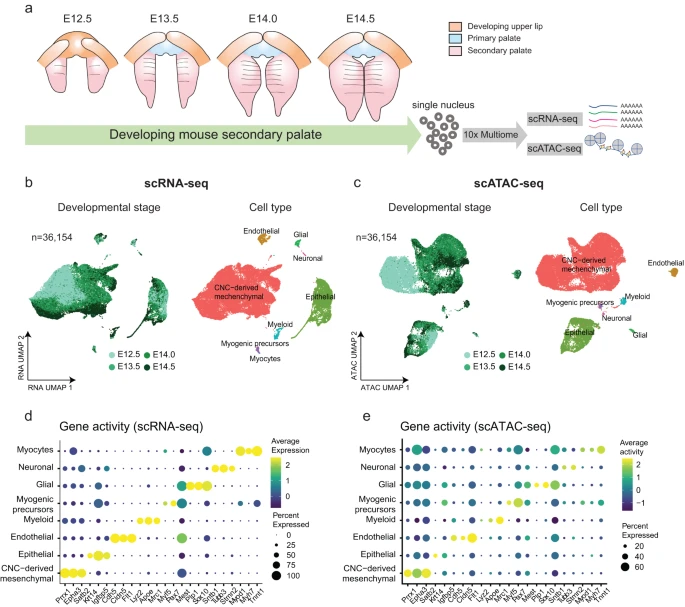
Here, we perform single-cell multiome sequencing and profile chromatin accessibility and gene expression simultaneously within the same cells (n = 36,154) isolated from mouse secondary palate across embryonic days (E) 12.5, E13.5, E14.0, and E14.5. We construct five trajectories representing continuous differentiation of cranial neural crest-derived multipotent cells into distinct lineages. By linking open chromatin signals to gene expression changes, we characterize the underlying lineage-determining transcription factors.
In silico perturbation analysis identifies transcription factors SHOX2 and MEOX2 as important regulators of the development of the anterior and posterior palate, respectively. In conclusion, our study charts epigenetic and transcriptional dynamics in palatogenesis, serving as a valuable resource for further cleft palate research. Reference